نظریه تابعی چگالی یک روش مدلسازی مکانیکی کوانتومی محاسباتی است که در علم فیزیک، کیمیا و علوم مواد مورد استفاده قرار میگیرد تا ساختار الکترونیکی(یا ساختار هستهای) (اصولاً حالت اولیه) بادی(body) – سیستمهای بسیاری از اجسام، بهویژه اتمها، مولکولها و مراحل تغلیذشده مواد را در تحقیقات بررسی کند. در این نظریه، با معرفی تابعی جهان شمول انرژی و وردش گیری از آن، ویژگیهای الکترونی ماده (در اینجا چگالی الکترون) بدست میآید.
با استفاده از این تئوری، میتوان با استفاده از تابعی(تابعیت)، یعنی توابع یک تابع دیگر، خصوصیات یک سیستم الکترون را تعیین کرد، که در این حالت مکانی الکترون به چگالی الکترون وابسته است. از این رو، تئوری تابعی چگالی نام ناشی از استفادهای تابعی یا چگالی الکترونها است. این نظریه، از جمله رایجترین و محبوبترین روشهای موجود در فیزیک ماده چگال، فیزیک محاسباتی و کیمیای محاسباتی است.
این نظریه ریشه در مدل توماس – فرمی دارد، و برپایه دو قضیه هوهنبرگ – کوهن بنا شدهاست. توضیح پدیدههایی مانند نیروهای بینمولکولی، به ویژه نیروی واندروالسی، نوار ممنوعه در نیمهرساناها، انتقال بار در حالت برانگیخته و... با این روش بطور کامل امکانپذیر نیست و پژوهش برای ایجاد تغییراتی که این محدودیتها را از بین ببرند ادامه دارد.
نظریه تابعی چگالی از سال ۱۹۷۰ یکی از محبوبترین روشها فیزیک حالت جامد بودهاست. با این حال تا سال ۱۹۹۰ که تقریبهای در نظر گرفته شده در تئوری آن مورد تجدید نظر قرار گرفت و مدل بهتری برای برهمکنشهای تبادلی ارائه شد، به عنوان یک روش دقیق در شیمی کوانتومی در نظر گرفته نشد. هزینههای محاسباتی در مقایسه با روشهای سنتی، مانند تبادل، فقط تئوری هارتری – فوک و فرزندان آن که همبستگی الکترونی را شامل میشوند، نسبتاً کم هستند.
با وجود پیشرفتهای اخیر، هنوز هم در استفاده از نظریه تابعی چگالی برای توصیف صحیح، مشکلاتی وجود دارد. تعامل بین مولکولی (از اهمیت اساسی در درک واکنش های شیمیایی)، بهویژه نیروهای واندروالس(پراکندگی)؛ حالت برانگیخته انتقال چارج؛ حالت گذار، سطوح انرژی پتانسیل جهانی، تعامل ناگهانی و برخی از سیستمهای بهشدت همبسته؛ و در محاسبات گاف انرژی و فرومقناطیس در نیمههادیها این مشکلات قابل مشاهده است. درمان ناقص پراکندگی میتواند بر دقت DFT (حداقل در صورت استفاده به تنهایی و اصلاح نشده)، در درمان سیستمهاییکه تحت تأثیر پراکندگی قرار دارند (به عنوان مثال در تعامل اتمهای گاز نجیب) یا جاییکه پراکندگی با سایر اثرات (بهعنوان مثال در مولکولهای زیستی) تحت تأثیر قرار میگیرد، تأثیر منفی بگذارد. توسعه روشهای جدید DFT که برای رفع این مشکل ایجاد شده است، با تغییراتی در عملکردها یا با درج اصطلاحات افزودنی، یک موضوع تحقیقاتی کنونی است.
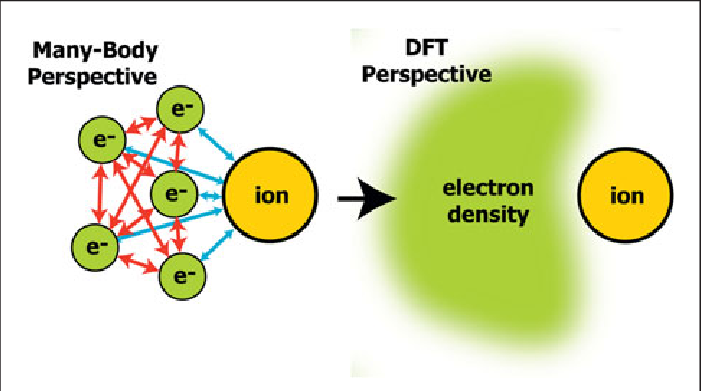
مرور کلی روش
در زمینه علم مواد محاسباتی، محاسبات اولیه ab initio (از اصول اول) محاسبه DFT اجازه میدهد پیشبینی و محاسبه رفتار مواد بر اساس ملاحظات مکانیکی کوانتومی، بدون نیاز به پارامترهای مرتبه بالاتر مانند خصوصیات اساسی ماده صورت گیرد. در تکنیکهای DFT معاصر ساختار الکترونیکی با استفاده از تابع پتانسیلی بر روی الکترونهای سیستم ارزیابی میشود. این پتانسیل DFT بهعنوان مجموع پتانسیلهای خارجی Vext ساخته میشود، که فقط با ساختار و ترکیب عنصری سیستم و یک پتانسیل موثر Veff که نمایانگر تعاملات بین الکترونیکی است، تعیین میشود. بنابراین، یک مشکل برای یک سوپرسل یک ماده با n الکترون میتواند بهعنوان مجموعهای از n معادله یک الکترونی شرودینگر، که بهعنوان معادلات کوهن – شم نیز شناخته می شوند، مطالعه شود.
سرچشمههای نظریه تابعی چگالی
اگرچه نظریه تابعی چگالی ریشه در مدل توماس – فرمی برای ساختار الکترونیکی مواد دارد، اما DFT برای اولینبار توسط والتر کوهن و پیر هوهنبرگ در چارچوب دو قضیه هوهنبرگ – کوهن(H-K) در یک پایه تئوریک محکم قرار گرفت. قضایای اصلی H-K فقط در صورت عدموجود میدان مقناطیسی فقط برای حالتهای غیر انحطاطی در سطح زمین برگزار میشود، اگرچه از آن زمان برای این موارد تعمیم یافته اند.
قضیه H-K اول نشان میدهد که خصوصیات حالت پایه یک سیستم چندالکترونی بسیار خاص توسط یک چگالی الکترونیکه تنها به سه مختصات مکانی بستگی دارد، تعیین میشوند. این کار زمینه را برای کاهش مشکل چندجسمی N الکترون با مختصات مکانی N3 به سه مختصات مکانی از طریق استفاده از نظریه تابعی چگالی الکترون فراهم کرد. این قضیه از آن زمان به دامنه وابسته به زمان گسترش یافته است تا نظریه تابعی چگالی وابسته به زمان (TDDFT) توسعه یابد، که میتواند برای توصیف حالات برانگیخته استفاده شود.
قضیه H-K دوم یک انرژی تابعی برای سیستم تعریف میکند و اثبات میکند که چگالی الکترونی صحیح حالت پایه این انرژی تابعی را به حداقل می رساند.
در کارهاییکه بعدا آنها جایزه نوبل کیمیا را بهدست آورد، قضیه H-K توسط والتر کوهن و لو جیو شم برای تولید Kohn-Sham DFT (KS DFT) ساخته شد. در این چارچوب، مشکل غیرقابل تحمل چند جسمی الکترونهای متقابل در پتانسیل خارجی استاتیک، به یک مشکل قابل انتقال الکترونهای غیر متقابل در حال حرکت در یک پتانسیل مؤثر کاهش مییابد. پتانسیل مؤثر شامل پتانسیل خارجی و اثرات متقابل کولومب بین الکترونها، به عنوان مثال، تعامل تبادلی و همبستگی(exchange – correlation reactions) است. مدلسازی دو تعامل اخیر در KSDFT به مشکل مواجه میشود. سادهترین تقریب، تقریب چگالی موضعی(LDA) است، که بر اساس مبادله انرژی دقیق برای یک گاز الکترونی یکنواخت است، که میتواند از مدل توماس – فرمی، و از متناسب کردن با انرژی همبستگی برای یکگاز الکترونی یکنواخت بهدست میآید. حل سیستمهای غیرتعاملی نسبتا آسان هستند زیرا عملکرد موج میتواند بهعنوان تعیینکننده Slater از مدارها نمایان شود. علاوه براین، انرژی جنبشی تابعی چنین سیستمی دقیقاً شناخته شده است. بخش تبادلی – همبستگی از کل تابعی انرژی ناشناخته است و باید تقریب یابد.
یک رویکرد دیگر، که کمتر از KSDFT محبوبیت دارد اما احتمالاً از نزدیک با روح قضایای اصلی H-K ارتباط دارد، نظریه تابعی چگالی بدون اربیتال(OFDFT) است، که در آن از تابعیتهای تقریبی نیز برای انرژی جنبشی سیستم غیرتعاملی استفاده میشود.
برنامههای کاربردی برای DFT
بهطور کلی، نظریه تابعی چگالی کاربردهای فزایندهای در کیمیا و علوم مواد برای تفسیر و پیشبینی رفتار سیستم پیچیده در مقیاس اتمی پیدا میکند. بهطور خاص، روشهای محاسباتی DFT برای سیستمهای مرتبط با سنتز و پارامترهای پردازش استفاده میشود. در چنین سیستمهایی، مطالعات تجربی غالباً با نتایج متناقض و شرایط غیر تعادل اشتباه میشوند. نمونههایی از کاربردهای DFT معاصر شامل مطالعه تأثیر دوپانتها(dopants) بررفتار تحول فاز در اکسایدها، رفتار مقناطیسی در مواد نیمههادی مقناطیسی رقیق و مطالعه رفتار مقناطیسی و الکترونیکی در فروالکتریکها و نیمههادیهای مقناطیسی رقیق است. همچنین نشان داده شده است که DFT در پیشبینی حساسیت برخی از نانوساختارها به آلایندههای محیطی مانند دای اکساید گوگرد یا اکولولین و همچنین پیشبینی خواص مکانیکی نتایج خوبی میدهد.
در عمل، تئوری کوهن – شم بسته بهآنچهکه مورد بررسی قرار میگیرد، میتواند از چند طریقهای متفاوت استفاده شود. در محاسبات حالت جامد، تقریب چگالی موضعی معمولاً به همراه مجموعههای پایهموج تخت مورد استفاده قرار میگیرد، زیرا یک رویکرد گاز الکترونی برای الکترونهاییکه از طریق جامد بینهایت جابجایی میشوند مناسبتر است. در محاسبات مولکولی، تابعیتهایی پیشرفته تر مورد نیاز است، و تنوع عظیمی از تابعیت همبستگی تبادل شده برای کاربردهای کیمیایی توسعه یافته است. برخی از اینها با تقریب گاز یکنواخت الکترون مغایر است. با اینحال، آنها باید به LDA در حد گاز الکترون کاهش یابند. در بین فیزیکدانان، یکی از تابعیتهای پرکاربرد، مدل تبادل شده Perdew-Burke-Ernzerhof است(یک پارامترسازی شیب مستقیم تعمیمیافته از گاز الکترونی آزاد و بدون پارامترهای آزاد). با اینحال، این برای محاسبات مولکولی فاز گاز به اندازه کافی از نظر کالریکی دقیق نیست. در جامعه کیمیا، یکی از کارکردهای محبوب با عنوان BLYP (از نام Becke برای بخش مبادله و لی، یانگ و پار برای بخش همبستگی) شناخته شده است. حتی بیشتر مورد استفاده B3LYP است، که یک تابعیت هیبریدی است که در آن انرژی مبادله، از عملکرد تبادلی بک(Becke)، با انرژی دقیق از نظریه هارتری – فوک ترکیب میشود. همراه با تبادل مؤلفهها و تابعیت همبستگی، سه پارامتر تابعیت هیبریدی(ترکیبی) را مشخص میکنند، و مشخص میکنند که مبادله دقیق چه مقدار در آن مخلوط شده است. اگرچه نتایج بهدست آمده با این تابعیتها(Functional) برای اکثر برنامهها معمولاً بهاندازه کافی دقیق است، اما هیچ روش منظمی برای بهبود آنها وجود ندارد(برخلاف برخی از روشهای سنتی مبتنی بر تابع موج، مانند تعامل پیکربندی یا نظریه خوشه همراه). در رویکرد فعلی DFT نمیتوان خطای محاسبات را بدون مقایسه آنها با سایر روشها یا آزمایشات تخمین زد.
شبه پتانسیلها Pseudo-potentials
اگر الکترونها بهدو گروه تقسیم شوند، الکترون ولانسی و الکترونهای مغزی، معادله الکترون شرودینگر میتواند بسیار ساده شود. الکترونهای موجود در پوستههای داخلی کاملاً محدود بوده و نقش مهمی در اتصال کیمیایی اتمها ندارند. آنها همچنین تا حدودی هسته را پوشش غربالی میدهند، بنابراین یک هسته تقریبا بیاثر شکل میگیرد. خواص اتصال تقریبی کاملا ناشی از الکترونهای ظرفیتی(ولانسی) است، خصوصا در فلزات و نیمه هادیها. این جدایی نشان میدهد که الکترونهای داخلی را میتوان در تعداد زیادی از موارد نادیده گرفت و از این طریق اتم را بهیک هسته یونی کاهش داد که با الکترونهای ولانسی در تعامل است. استفاده از یک تعامل مؤثر، یک شبه پتانسیل، که تقریباً پتانسیل احساس شده توسط الکترون های والانس را تقریب میکند، برای اولین بار توسط فرمی در سال 1934 و هلمن در سال 1935 پیشنهاد شد.
پاشش الکترونی Electron Smearing
الکترونهای یک سیستم کمترین حالتهای خاص کوهن – شم را تا یک سطح انرژی معین با توجه به اصل اُفباو Aufbau اشغال خواهند کرد. این مربوط به توزیع مرحله مانند فرمی – دیراک در صفر مطلق است. اگر چندین حالت تخریب شده یا نزدیک به ویژهحالتهای تخریب شده در سطح فرمی وجود داشته باشد، میتوان مشکلات همگرایی را پیدا کرد، زیرا آشفتگیهای بسیار کوچک ممکن است اشغال الکترون را تغییر دهد.
یکی از راه های میرایی این نوسانات، لکهدار کردن الکترون هاست، یعنی اجازه اشغال کسری. یک روش برای انجام این کار، اختصاص درجه حرارت محدود به توزیع الکترومی فرمی – دیراک است. روشهای دیگر مختص به توزیع گاوسی تجمعی الکترونها یا استفاده از روش Methfessel-Paxton است.
ترجمه: آصف برخیا - آ بی کلاس
+ نوشته شده در پنجشنبه ۱۳۹۸/۰۵/۳۱ ساعت 0:7 توسط آصف ابراهیمی
|




 همه ای ما کمابیش لذت درک کردن چیزی را چشیده ایم؛ لذتی که در تک تک سلول های مغزمان جاری می شود و تمام وجودمان را فرا می گیرد. با درک کردن و فهمیدن، احساس قدرت می کنیم. گاهی ممکن است چیزی را نفهمیم؛ آری زیاد پیش می آید. گاهی هم می پنداریم چیزی را فهمیده ایم و به آسانی از کنارش رد می شویم. اما گاهی مصرانه باقی می مانیم وکم نمی آوریم؛ آنقدر پافشاری می کنیم تا به واقعیت دست یابیم. اما چه زمان است که چیزی رامی فهمیم؟ اصلا فهمیدن به چه معنی است وتفاوت بین یادگیری و فهمیدن در چیست؟
همه ای ما کمابیش لذت درک کردن چیزی را چشیده ایم؛ لذتی که در تک تک سلول های مغزمان جاری می شود و تمام وجودمان را فرا می گیرد. با درک کردن و فهمیدن، احساس قدرت می کنیم. گاهی ممکن است چیزی را نفهمیم؛ آری زیاد پیش می آید. گاهی هم می پنداریم چیزی را فهمیده ایم و به آسانی از کنارش رد می شویم. اما گاهی مصرانه باقی می مانیم وکم نمی آوریم؛ آنقدر پافشاری می کنیم تا به واقعیت دست یابیم. اما چه زمان است که چیزی رامی فهمیم؟ اصلا فهمیدن به چه معنی است وتفاوت بین یادگیری و فهمیدن در چیست؟